


Клинический разбор в общей медицине №01 2026
Клинический случай наследственного ангиоотека
Номера страниц в выпуске:27-30
Аннотация
Первичные иммунодефицитные состояния, как генетически обусловленные заболевания, имеют различные варианты клинических проявлений и порой скрываются под масками других заболеваний. Наследственный ангиоотек (НАО) является орфанным генетически обусловленным заболеванием и возникает как следствие снижения синтеза С1-ингибитора (С1-ИНГ) и/или снижения его функциональной активности. Отличительной особенностью НАО является сочетание отечности мягких тканей головы, верхних дыхательных путей и слизистой оболочки желудочно-кишечного тракта или возникновение изолированного отека последней. Провоцирующими факторами возникновения НАО являются стрессовые состояния, медицинские инвазивные манипуляции, при этом продолжительность клинических проявлений составляет от 2 до 5 сут. Снижение синтеза или функциональной активности С1-ИНГ приводит к накоплению брадикинина. Именно это и является причиной отсутствия эффекта от введения эпинефрина, системных глюкокортикостероидов, антигистаминных препаратов, что ухудшает прогноз заболевания и приводит к возникновению угрожающего жизни пациента состояния. НАО относится к разряду заболеваний с гиподиагностикой, с отсутствием настороженности у врачей первичного звена здравоохранения, а также, в случае изолированной абдоминальной атаки, может приводить к необоснованным хирургическим вмешательствам. В статье на примере клинического случая пациента с НАО, выявленного в Омской области, дана характеристика различных вариантов клинических проявлений НАО, продемонстрирована роль оценки наследственного анамнеза и триггеров НАО, подчеркнуто значение своевременно начатой терапии и рассмотрены современные возможности оказания медицинской помощи данной категории пациентов для спасения их жизни и улучшения ее качества.
Ключевые слова: наследственный ангиоотек, первичный иммунодефицит, брадикинин, ингибитор, комплемент, профилактика.
Для цитирования: Надей Е.В., Лепехина Е.С., Усачева Е.В. Клинический случай наследственного ангиоотека. Клинический разбор в общей медицине. 2026; 7 (1): 27–30. DOI: 10.47407/kr2026.7.1.00747
Первичные иммунодефицитные состояния, как генетически обусловленные заболевания, имеют различные варианты клинических проявлений и порой скрываются под масками других заболеваний. Наследственный ангиоотек (НАО) является орфанным генетически обусловленным заболеванием и возникает как следствие снижения синтеза С1-ингибитора (С1-ИНГ) и/или снижения его функциональной активности. Отличительной особенностью НАО является сочетание отечности мягких тканей головы, верхних дыхательных путей и слизистой оболочки желудочно-кишечного тракта или возникновение изолированного отека последней. Провоцирующими факторами возникновения НАО являются стрессовые состояния, медицинские инвазивные манипуляции, при этом продолжительность клинических проявлений составляет от 2 до 5 сут. Снижение синтеза или функциональной активности С1-ИНГ приводит к накоплению брадикинина. Именно это и является причиной отсутствия эффекта от введения эпинефрина, системных глюкокортикостероидов, антигистаминных препаратов, что ухудшает прогноз заболевания и приводит к возникновению угрожающего жизни пациента состояния. НАО относится к разряду заболеваний с гиподиагностикой, с отсутствием настороженности у врачей первичного звена здравоохранения, а также, в случае изолированной абдоминальной атаки, может приводить к необоснованным хирургическим вмешательствам. В статье на примере клинического случая пациента с НАО, выявленного в Омской области, дана характеристика различных вариантов клинических проявлений НАО, продемонстрирована роль оценки наследственного анамнеза и триггеров НАО, подчеркнуто значение своевременно начатой терапии и рассмотрены современные возможности оказания медицинской помощи данной категории пациентов для спасения их жизни и улучшения ее качества.
Ключевые слова: наследственный ангиоотек, первичный иммунодефицит, брадикинин, ингибитор, комплемент, профилактика.
Для цитирования: Надей Е.В., Лепехина Е.С., Усачева Е.В. Клинический случай наследственного ангиоотека. Клинический разбор в общей медицине. 2026; 7 (1): 27–30. DOI: 10.47407/kr2026.7.1.00747
Clinical case of hereditary angioedema
Elena V. Nadey, Ekaterina S. Lepekhina, Elena V. Usacheva1 Omsk State Medical University, Omsk, Russia;
2 V.P. Bisyarina City Children's Clinical Hospital No. 2, Omsk, Russia
ElenaV.Usacheva@yandex.ru
Abstract
Primary immunodeficiency conditions, as genetically determined diseases, have various clinical manifestations and sometimes hide under the masks of other diseases. Hereditary angioedema (HAE) is an orphan genetic disorder that occurs as a result of reduced synthesis of C1-inhibitor (C1-INH) and/or reduced functional activity of C1-INH. A distinctive feature of the HAE is characterized by the localization of edema not only in the head, neck, and upper respiratory tract, but by a combination or isolated edema of the gastrointestinal tract. The provoking factor in the appearance of edema is stressful conditions, medical invasive manipulations, while the duration of edema can last from 2 to 5 days. Decreased synthesis or functional activity of the C1-INH leads to the accumulation of bradykinin. This is the reason for the lack of effect from the administration of epinephrine, systemic glucocorticosteroids, antihistamines, which worsens the prognosis of the disease and acts as a life-threatening condition. HAE belongs to the category of diseases with underdiagnosis, with a lack of alertness among primary care doctors, and also, in the case of an isolated abdominal attack, leads to unjustified surgical interventions. The article, using the example of a clinical case of a patient with HAE identified in the Omsk region, characterizes various variants of clinical manifestations of HAE, demonstrates the role of assessing hereditary anamnesis and edema triggers, emphasizes the importance of timely therapy and modern possibilities of providing medical care to this category of patients to improve the quality and save their lives.
Keywords: hereditary angioedema, primary immunodeficiency, HAE, bradykinin, inhibitor, complement, prevention.
For citation: Nadey E.V., Lepekhina E.S., Usacheva E.V. Clinical case of hereditary angioedema. Clinical review for general practice. 2026; 7 (1): 27–30 (In Russ.). DOI: 10.47407/kr2026.7.1.00747
Введение
Наследственный ангиоотек (НАО) – орфанное генетически обусловленное заболевание с аутосомно-доминантным типом наследования, основными клиническими проявлениями которого выступают рецидивирующие ангиоотеки различной локализации с вовлечением слизистых оболочек и глубоких слоев кожи [1]. Распространенность НАО составляет примерно 1 случай на 50 тыс. человек [2].
НАО возникает в результате мутаций гена, кодирующего С1-ингибитор (С1-ИНГ). Существуют два основных типа НАО, которые наследуются по аутосомно-доминантному типу: НАО 1-го типа составляет примерно 85% случаев и возникает в результате количественного дефицита С1-ИНГ; НАО 2-го типа является причиной примерно 15% случаев и является следствием дисфункции белка С1-ИНГ [3]. Выделяют также форму НАО с нормальным уровнем С1-ИНГ.
Типичные признаки и симптомы НАО появляются в детстве или в период полового созревания и сохраняются на протяжении всей жизни. Дебют НАО в течение первых трех лет жизни отмечается лишь в 35% случаев, а у 65% пациентов приходится на возраст от 12 лет и старше. К сожалению, имеет место гиподиагностика заболевания: в среднем проходит 8–20 лет от момента возникновения симптомов до установления диагноза НАО [4].
Для НАО характерны рецидивирующие отеки различной локализации. Могут поражаться несколько областей тела, включая лицо, язык, гортань, дистальные отделы конечностей, стенку кишечника, гениталии. Приступы длятся от 72 до 96 ч, часто протекая в тяжелой форме, значительно снижая дееспособность и качество жизни пациентов, и при отсутствии оказания медицинской помощи могут приводить к летальному исходу. Обычно приступы начинаются с продромального ощущения покалывания, которое может сопровождаться незудящей сыпью. Тип ангиоотека, наблюдаемый при НАО, не связан с наличием крапивницы и не поддается лечению системными глюкокортикостероидами (ГКС) и/или антигистаминными препаратами (АГП), так как обусловлен эффектами брадикинина.
Для НАО характерны медленное нарастание отека и быстрое развитие асфиксии с момента появления симптомов нарушения дыхания (в среднем в течение 20–40 мин). Приступы такого типа встречаются редко, но более половины больных c НАО имеют хотя бы один орофарингеальный приступ в течение жизни, который может иметь фатальный исход [5].
Сложнее всего диагностировать НАО при изолированных абдоминальных отеках. Абдоминальные атаки возникают у 43–93% пациентов с НАО (по некоторым данным, до 70–80% случаев). До 80% пациентов имеют рецидивирующий характер абдоминальных приступов. На протяжении многих лет абдоминальные симптомы могут быть единственным симптомом НАО. Боли в животе описываются пациентами как «спазмы», «колики» и оцениваются как тяжелые, мучительные у 87% пациентов. Частыми симптомами выступают рвота, диарея, возникающие как проявление транзиторного отека стенки тонкого кишечника, приводящего к частичной или полной кишечной непроходимости, что обусловливает неотложную госпитализацию в хирургический стационар [6]. Частота и тяжесть атак непредсказуемы и различны у каждого пациента и так же непредсказуемо могут меняться во времени: в 28% случаев – каждые 2–4 мес, в 18% – раз в 2–3 мес, в 18% – реже чем каждые 6 мес, в 34% – более 1 раза в месяц.
Диагноз НАО устанавливается на основании тщательного сбора анамнеза заболевания, наследственного анамнеза, клинического обследования, анализа иммунологических показателей крови, выявляющего снижение уровня белков комплемента, и, в случаях высокого клинического подозрения и рецидивирующего ангиоотека неопределенной этиологии, генетического тестирования. Основной метод диагностики НАО – генетическое исследование, дающее возможность выявить мутации, значимые для развития НАО, и точно установить диагноз (особенно при НАО 3-го типа). Кроме того, выявленные мутации позволяют прогнозировать вероятность развития НАО на доклинической стадии. Также стоит отметить, что генетическое исследование более значимо для детей первого года жизни, поскольку исследование системы комплемента в этом возрасте нецелесообразно. В России разработан скрининг НАО, он касается родителей, братьев, сестер, детей пациентов с НАО и включает исследование уровня С4-компонента комплемента, исследование концентрации и функциональной активности С1-ИНГ с последующим (при наличии показаний) молекулярно-генетическим обследованием.
При установлении диагноза НАО каждый пациент должен быть обеспечен препаратом неотложной помощи – антагонистом В2-рецепторов брадикинина, который препятствует сцеплению излишне выработавшегося брадикинина с В2-рецепторами, для купирования приступов НАО, а также использования с целью обеспечения лечебного и профилактического лечения перед всевозможными диагностическими и лечебными травматическими медицинскими манипуляциями, в том числе оперативными, стоматологическими, гинекологическими и др. [7]. В стационарных условиях для купирования острых атак используют концентрат ингибитора С1-эстеразы. В экстренных случаях, при недоступности антагониста В2-рецепторов брадикинина и концентрата ингибитора С1-эстеразы, для оказания неотложной помощи пациентам с НАО используют плазму крови, представляющую собой донатор ингибитора С1-эстеразы. Аминокапроновая кислота используется у больных НАО off-lable, так как является единственным ингибитором фибринолиза для внутривенного введения. Ингибиторы фибринолиза могут быть назначены пациентам с НАО для купирования периферических атак и невыраженных абдоминальных атак.
С целью улучшения качества медицинской помощи пациентам с НАО при выявлении заболевания на каждого пациента оформляется «Паспорт пациента», где описываются порядок ведения пациента и алгоритм оказания неотложной помощи. Однако при поступлении пациента в стационар с клинической картиной абдоминальной атаки под маской острой кишечной непроходимости или острого ларингофарингеального отека «Паспорт пациента» специалистами зачастую игнорируется, что может привести к необоснованным хирургическим вмешательствам и даже смерти пациента.
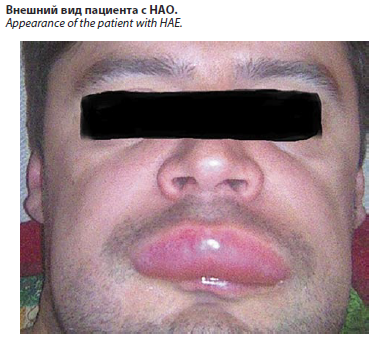 Клинический случай
Клинический случайПациент У., 24 лет, впервые обратился на консультацию к врачу аллергологу-иммунологу с жалобами на высыпания в области туловища в виде «лепешек» с небольшим зудом, появление плотных белых отеков в области лица, шеи с затруднением дыхания (см. рисунок), отечностью дистальных отделов нижних конечностей. При сборе анамнеза установлено, что отечность сохранялась у пациента до 3–4 сут, отсутствовало влияние проводимой терапии блокаторами гистаминовых рецепторов и системными ГКС на частоту, выраженность и длительность клинических проявлений НАО. Причину появления отечности мягких тканей и высыпаний четко связать с чем-либо не удалось, пищевую и лекарственную непереносимость пациент отрицает. Наследственный анамнез не отягощен.
Из анамнеза заболевания известно, что с 10-летнего возраста ежегодно, независимо от сезона, без видимой причины отмечались высыпания на теле и отеки в области лица, наблюдался у педиатра с приемом АГП по потребности. В подростковом возрасте изменилась локализация отеков – они стали появляться в области стоп или кистей, чаще всего после физической нагрузки. В большинстве случаев отеки возникали без видимой причины, иногда отмечал, что провоцирующим фактором могла быть физическая нагрузка или механическое воздействие на кожу (травма). Эпизодически отмечал высыпания на теле в виде красных пятен, не сопровождающихся зудом, без четкой связи с приемом пищи или действием каких-либо факторов. Продолжал наблюдение у участкового педиатра.
С возрастом частота появления отечности возрастала, эффекта от приема АГП не отмечал. Отечность развивалась в течение суток и чаще всего проходила самостоятельно через 3–4 дня. Период с момента начала заболевания до обращения к врачу аллергологу-иммунологу составил 14 лет.
В 2002 г., в возрасте 24 лет, пациент впервые обратился на прием к врачу аллергологу-иммунологу. Было проведено общеклиническое обследование и выявлены следующие изменения: гиперхолестеринемия (8,05 ммоль/л); ультразвуковое исследование – застой желчи в желчном пузыре, добавочная селезенка 17 мм; фиброгастродуоденоскопия – явления катарального гастрита и бульбита, рубцовая деформация луковицы двенадцатиперстной кишки; фиброколоноскопия – хронический колит; электрокардиография – метаболические изменения в миокарде. Пациенту назначено лечение в виде трех курсов неспецифической гипосенсибилизирующей терапии стабилизаторами мембран тучных клеток с улучшением в виде купирования высыпаний, однако отеки дистальных отделов преимущественно нижних конечностей продолжали беспокоить с периодичностью 1–2 раза в месяц, эффекта от приема АГП и системных ГКС по-прежнему не отмечалось.
С 2005 г. пациент впервые стал замечать, что до появления отечности возникали боль в эпигастральной области (на фоне стрессов) и высыпания в области туловища (не всегда), только затем, через час, появлялась отечность в области горла и дистальных отделов конечностей. Стала прослеживаться связь появления отеков с физическим перенапряжением или волнением. Кроме того, появились данные наследственного анамнеза – у пациента за эти годы родился сын, у которого возникали эпизоды отечности в области гортани. В период очередной госпитализации в отделение аллергологии-иммунологии пациенту было проведено иммунологическое обследование: уровень С3-компонента комплемента – 0,2 г/л (при норме 0,9–18,0 г/л), С4-компонента комплемента – 0,34 г/л (при норме 0,16–0,54 г/л). Выявленное снижение уровня С3-компонента комплемента, с учетом появления данных наследственного анамнеза (ангиоотеки у сына), особенностей отечности и связи с физической нагрузкой или стрессовыми ситуациями, отсутствия эффекта от проводимой терапии АГП и системными ГКС, косвенно подтверждало наследственный характер ангионевротического отека. От проведения генетического обследования в Федеральном центре пациент воздержался. Для долгосрочной профилактики был назначен даназол в дозе 200 мг/сут в течение 3 мес, затем пациент был переведен на терапию в дозе 100 мг/сут. Однако число периферических отеков, эпизодов отечности гортани достигало кратности 1–2 раза в месяц с необходимостью обращения за неотложной медицинской помощью; появились рецидивирующие фурункулы, гидраденит.
В 2007 г. с клинической картиной выраженного болевого синдрома в области живота пациент был доставлен бригадой скорой медицинской помощи в приемное отделение стационара, где было проведено хирургическое вмешательство – диагностическая лапароскопия, по результатам которой патологический очаг не выявлен; в дальнейшем переведен в отделение реанимации с подозрением на острый панкреатит. После дообследования диагноз острого панкреатита не подтвердился, за время наблюдения рецидивировали отеки в области бедра, гортани, мошонки. С этого момента отеки в области гортани или конечностей всегда сопровождались абдоминальными болями.
С 2007 г. пациент был переведен на долгосрочную профилактику с использованием антифибринолитического средства (транексамовая кислота в средней дозе 1,0–1,5 г/сут) под контролем свертывающей системы крови. Частота приступов уменьшилась до 1 раза в 6–8 нед. В «Паспорт больного наследственным ангиоотеком для пациентов старше 18 лет» были внесены рекомендации по оказанию помощи в неотложных ситуациях, в период атаки, в случае хирургических вмешательств или инвазивных манипуляций.
В 2008 г. появились новые данные наследственного анамнеза – у матери пациента впервые возник отек в области правой ушной раковины. При проведении иммунологического исследования у нее было выявлено снижение уровня С3-компонента комплемента до 0,04 г/л.
В последующем, после однократного эпизода ангиоотека, рецидивы отеков не отмечались.
В 2014 г., в возрасте 36 лет, на основании проведенного генетического исследования пациент был включен в региональный сегмент Федерального регистра лиц, страдающих жизнеугрожающими и хроническими прогрессирующими редкими (орфанными) заболеваниями. В качестве неотложной терапии для купирования жизнеугрожающих атак НАО был назначен блокатор брадикининовых В2-рецепторов.
В 2015 г. число абдоминальных атак резко снизилось, прием транексамовой кислоты осуществлялся по потребности не чаще 4 раз в год. В настоящее время два сына пациента с подтвержденным диагнозом НАО также включены в Федеральный регистр лиц, страдающих жизнеугрожающими и хроническими прогрессирующими редкими (орфанными) заболеваниями.
Описанный случай НАО, несомненно, представляет клинический интерес и демонстрирует особенности течения НАО, который проявился в трех поколениях.
Заключение
НАО представляет собой серьезную медико-социальную проблему, так как влияет на качество жизни пациентов и может привести к летальному исходу при отсутствии своевременного оказания неотложной помощи и адекватной профилактической терапии. Поздняя диагностика заболевания, принятие неверных решений в тактике ведения пациентов с НАО, высокий риск неблагоприятных исходов заболевания обусловлены:
• низкой настороженностью врачей первичного звена в силу относительно невысокой частоты встречаемости НАО;
• недостаточно качественным сбором анамнеза, в том числе наследственного (особенно при наличии ближайших родственников с симптомами НАО в виде рецидивирующих отеков, ургентных ситуаций, связанных с нарушением дыхания / асфиксией, трахеотомиями, эпизодами «острого живота»);
• отсутствием анализа триггеров атак НАО, таких как стресс, нагрузки, диагностические и лечебные медицинские манипуляции;
• многообразием клинических проявлений НАО, в том числе изолированных абдоминальных атак, поскольку исключить и даже заподозрить НАО в случае первой абдоминальной атаки без развития периферических ангиоотеков практически невозможно;
• недостаточным вниманием к отсутствию эффекта от проведения терапии АГП, системными ГКС.
В связи с указанными трудностями следует подчеркнуть необходимость обучения врачей различных специальностей методам диагностики и лечения НАО, что поможет существенно улучшить выявляемость данного заболевания, от ранней диагностики которого зависят своевременное назначение эффективных (особенно для комплементзависимых ангиоотеков) лекарственных средств и качество жизни пациентов.
Конфликт интересов. Авторы заявляют об отсутствии конфликта интересов.
Conflict of interests. The authors declare that there is not conflict of interests.
Финансирование. Исследование не имело спонсорской поддержки.
Financing. The study had no sponsorship.
Список литературы доступен на сайте журнала https://klin-razbor.ru/
The list of references is available on the journal‘s website https://klin-razbor.ru/
Информация об авторах
Information about the authors
Надей Елена Витальевна – канд. мед. наук, доц., доц. каф. внутренних болезней и семейной медицины ДПО ФГБОУ ВО «Омский государственный медицинский университет». E-mail: nadeyelena@yandex.ru; ORCID: 0000-0003-0440-7118; Scopus Author ID: 56500711200
Elena V. Nadey – Cand. Sci. (Med.), Assoc. Prof., Omsk State Medical University. E-mail: nadeyelena@yandex.ru; ORCID: 0000-0003-0440-7118; Scopus Author ID: 56500711200
Лепехина Екатерина Сергеевна – врач аллерголог-иммунолог отд-ния аллергологии и иммунологии БУЗ ОО «ДГКБ №2 им. В.П. Бисяриной». E-mail: lepehina.ekaterina1995@gmail.com;
ORCID 0009-0003-0851-5367; Researcher ID: NYS-6564-2025
Ekaterina S. Lepekhina – allergologist-immunologist, V.P. Bisyarina State Clinical Hospital No. 2.
E-mail: lepehina.ekaterina1995@gmail.com;
ORCID 0009-0003-0851-5367; Researcher ID: NYS-6564-2025
Усачева Елена Владимировна – канд. мед. наук, доц., доц. каф. внутренних болезней и семейной медицины ДПО ФГБОУ ВО «Омский государственный медицинский университет».
E-mail: elenav.usacheva@yandex.ru; ORCID 0000-0002-6134-1533;
Researcher ID: I-4077-2017; Scopus Author ID: 56380398700
Elena V. Usacheva – Cand. Sci. (Med.), Assoc. Prof., Omsk State Medical University. E-mail: elenav.usacheva@yandex.ru;
ORCID 0000-0002-6134-1533; Researcher ID: I-4077-2017;
Scopus Author ID: 56380398700
Поступила в редакцию: 19.06.2025
Поступила после рецензирования: 09.07.2025
Принята к публикации: 17.07.2025
Received: 19.06.2025
Revised: 09.07.2025
Accepted: 17.07.2025
Список исп. литературыСкрыть список1. Sinnathamby ES, Issa PP, Roberts L et al. Hereditary Angioedema: Diagnosis, Clinical Implications, and Pathophysiology. Adv Ther 2023;40(3):814-27. DOI: 10.1007/s12325-022-02401-0
2. Maurer M, Magerl M, Ansotegui I et al. The international WAO/ EAACI guideline for the management of hereditary angioedema The 2017 revision and update. Allergy 2018;73(8):1575-96. DOI: 10.1111/all.13384
3. Zhang Y, Tortorici MA, Pawaskar D et al. Exposure-Response Model of Subcutaneous C1-Inhibitor Concentrate to Estimate the Risk of Attacks in Patients With Hereditary. Angioedema. CPT Pharmacometrics Syst Pharmacol 2018;7(3):158-65. DOI: 10.1002/psp4.12271
4. Banerji A, Li Y, Busse P et al. Hereditary angioedema from the patient's perspective: A follow-up patient survey. Allergy Asthma Proc 2018;39(3):212-23. DOI: 10.2500/aap.2018.39.4123
5. Bork K, Hardt J, Witzke G. Fatal laryngeal attacks and mortality in hereditary angioedema due to C1-INH deficiency. J Allergy Clin Immunol 2012;130(3):692-7. DOI: 10.1016/j.jaci.2012.05.055
6. Schöffl C, Wiednig M, Koch L et al. Hereditary angioedema in Austria: prevalence and regional peculiarities. J Dtsch Dermatol Ges 2019;17(4):416-23.
7. Maurer M, Aberer W, Caballero T et al. The Icatibant Outcome Survey: 10 years of experience with icatibant for patients with hereditary angioedema. Clin Exp Allergy 2022;52(9):1048-58. DOI: 10.1111/cea.14206
18 февраля 2026
Количество просмотров: 167