Клинический разбор в общей медицине №05 2026
1 Ural State Medical University, Yekaterinburg, Russia;
2 New Hospital LLC, Yekaterinburg, Russia
polina.tsaplina@yandex.ru
Abstract
Miyoshi myopathy belongs to the group of dysferlinopathies, rare myopathies, and therefore remains undiagnosed for a long time and currently has no etiopathogenetic treatment. The article presents a clinical case of Miyoshi myopathy in an 18-year-old patient who had an increase in creatine phosphokinase and hepatic transaminases for 3 years in the absence of subjective complaints. The article describes in detail additional diagnostic tests carried out for the purpose of differential diagnosis, as well as presents the data of medical genetic counseling.
Keywords: Miyoshi myopathy, dysferlinopathy, hypercreatinephosphatemia.
For citation: Poletaeva N.B., Teplyakova O.V., Tsaplina P.K. A case of Miyoshi myopathy in clinical practice. Clinical review for general practice. 2026; 7 (5): 133–137 (In Russ.). DOI: 10.47407/kr2026.7.5.00854
Введение
Дисферлинопатии – группа редких, фенотипически разнородных мышечных заболеваний. В основе дисферлинопатий лежит дефект синтеза дисферлина – белка, участвующего в репарации мембран мышечных клеток, поэтому следствием нарушения его синтеза является постепенная дегенерация скелетной мускулатуры [1–3].
В настоящее время описано как минимум 9 фенотипов дисферлинопатий, однако наиболее изучены четыре: миопатия Миоши типа 1 (MMD1), поясно-конечностная мышечная дистрофия рецессивного типа 2 (LGMDR2), дистальная миопатия с поражением передней большеберцовой кости (DMAT), бессимптомная гиперкреатинфосфаткиназемия. Все подтипы характеризуются аутосомно-рецессивным типом наследования, повышением уровня сывороточной креатинфосфокиназы (КФК) и, в большинстве случаев, слабостью определенных групп мышц [3, 4].
В настоящее время прогноз пациентов с дисферлинопатиями не определен, этиопатогенетическое лечение не разработано [3, 5–7].
Клинический случай
Пациент Б., 18 лет, студент, в феврале 2021 г. направлен на первичную консультацию к ревматологу в связи с изменениями в анализах крови в виде повышения уровня КФК более чем в 120 раз от верхней границы нормы (ВГН), а также аланинаминотрансферазы (АЛТ) и аспартатаминотрансферазы (АСТ) при отсутствии каких-либо субъективных жалоб.
Анамнез заболевания. Впервые повышение КФК было выявлено в 2019 г. в 17-летнем возрасте при прохождении «спортивной комиссии», ранее подобных исследований не проводилось. Обращала на себя внимание стойкость лабораторных изменений, кроме того, одновременно определялось повышение уровней АЛТ и АСТ.
В связи с установленными лабораторными изменениями в 2020 г. пациент был направлен на дообследование в гастроэнтерологическое отделение детской городской больницы с диагнозом «криптогенный гепатит».
По данным представленной медицинской документации, были проведены следующие лабораторные исследования:
• общеклинические анализы крови и мочи, копрограмма – без патологии.
• биохимический анализ крови: общий белок – 89,4 г/л; альбумины – 58,6% (52,4 г/л); глобулины: альфа-1 – 3,5%, альфа-2 – 7,8%, бета – 10%, гамма – 19,9%; мочевина – 3,8 ммоль/л; общий холестерин – 3,8 ммоль/л.
• HBsAg и anti-HCV – не обнаружены.
• тромбоэластограмма – нормокоагуляция в цельной крови.
• концентрация меди в сыворотке крови – 23,6 мкмоль/л (норма 11–22 мкмоль/л).
• церулоплазмин – 269,3 мг/л (норма 90–510 мг/л).
• анализ суточной мочи на медь – 0,0547 мг/сут (норма 0,0150–0,05 мг/сут).
• тандемная масс-спектрометрия – данных, свидетельствующих о наличии наследственных аминоацидопатий, органических ацидурий и дефектов окисления жирных кислот, не получено.
• IgA и IgG к деамидированным пептидам глиадина, эндомизию и тканевой трансглутаминазе – в пределах нормы.
Также были проведены следующие инструментальные исследования:
• ультразвуковое исследование органов брюшной полости – органической патологии не выявлено.
• эзофагогастродуоденоскопия: умеренно выраженный гастрит тела и антрального отдела желудка, дуоденогастральный рефлюкс, произведена биопсия слизистой оболочки двенадцатиперстной кишки.
• гистологическое исследование биопсийного материала слизистой оболочки двенадцатиперстной кишки – структура слизистой оболочки сохранена, атрофии и метаплазии нет, щеточная кайма сохранена; интраэпителиальный лимфоцитоз и нейтрофилез. Заключение: морфологическая картина соответствует классификации Marsh1, не исключается целиакия в сочетании с активным дуоденитом.
• пункционная биопсия печени под ультразвуковым контролем: в гепатобиоптате явления гиалиново-капельной белковой дистрофии гепатоцитов, умеренный портальный склероз, очаговая лимфогистиоцитарная инфильтрация, занимающая менее ⅓ портальных трактов, мелкое скопление мононуклеаров в паренхиме дольки. Заключение: хронический гепатит с перипортальным склерозом, минимальной активности. Индекс гистологической активности – 3 балла. Гель-инфузионная сонография – 1 балл. Окраска на медь и железо – отрицательно.
Таким образом, полученные данные не объясняли повышение уровней КФК и трансаминаз. Пациент был выписан под наблюдение гастроэнтеролога с рекомендациями приема эзомепразола в течение 1 мес и урсодезоксихолиевой кислоты в течение 6 мес.
Далее в течение года пациент неоднократно выполнял исследования как на фоне спортивных тренировок, так и без них. Максимальные значения КФК на фоне тренировок достигали уровня 31 000 Ед/л (норма до 180 Ед/л), в случае месячного перерыва от активных физических упражнений уровень КФК снижался до 9000 Ед/л, но по-прежнему более чем в 50 раз превышал ВГН. Одновременно неоднократно диагностировалось повышение уровней АЛТ и АСТ – до 5–6-кратного превышения ВГН.
Анамнез жизни. Единственный ребенок в семье. Начал заниматься фитнесом с 16 лет. Употребление каких-либо лекарственных средств, биологически активных и спортивных добавок пациент категорически отрицает. Биохимические исследования крови у родителей не выявили отклонений от нормы.
Данные объективного осмотра (апрель 2022 г.). Состояние удовлетворительное, температура тела 36,6°C. Кожа чистая, отеков нет. Периферические лимфатические узлы не изменены, щитовидная железа не пальпируется. Аускультативно в легких везикулярное дыхание, хрипы не выслушиваются. Тоны сердца ясные, ритм правильный, шумы отсутствуют, частота сердечных сокращений – 51 в минуту. Артериальное давление на обеих руках 120/70 мм рт. ст. Живот при пальпации мягкий, безболезненный. Печень по краю реберной дуги, селезенка не пальпируется. Мочеиспускание без изменений, стул регулярный, оформленный.
Status localis: суставы не деформированы, без экссудации, движения в полном объеме. Сила дистальных и проксимальных мышц сохранена.
Дополнительные методы исследования. С целью исключения эндокринологической и аутоиммунной патологии, в частности полимиозита, были проведены исследования крови на тиреотропный гормон, паратгормон, кальций, калий и натрий – результаты в пределах нормальных значений. Иммуноблот атинуклеарных антител – отрицательный результат.
Были проведены следующие инструментальные исследования:
• стимуляционная электронейромиография (ЭНМГ) – проведение не нарушено.
• игольчатая ЭНМГ передней большеберцовой мышцы справа, vastus medianus четырехглавой мышцы слева, дельтовидной мышцы слева: выявлены потенциалы двигательных единиц повышенной амплитуды и уменьшенной длительности, что можно расценивать как признак первично-мышечной патологии в стадии компенсации. Минимальная спонтанная активность в виде потенциалов фибрилляции (+) выявлена в четырехглавой и дельтовидной мышцах, что может свидетельствовать о минимально выраженном процессе мышечной дегенерации.
• биопсия четырехглавой мышцы бедра: в биоптате фрагменты скелетной мышцы. Поперечная исчерченность в основном сохранена. Соединительнотканные прослойки (эндомизий и перимизий) с мелкими очагами базофилии и незначительной лимфогистиоцитарной фокальной инфильтрацией с наличием плазмоцитов. Единичные фокусы с проникновением клеток инфильтрата в мышечное волокно с его повреждением глыбчатым распадом. Сосуды в основном не изменены, но в стенках отдельных артериол – единичные лимфоциты. В нескольких срезах инфильтрация более выражена и в лимфатических сосудах выявляются клеточные лимфогистиоцитарные «пробки» с примесью плазмоцитов. В эндомизии в зоне более выраженной инфильтрации определяются и тучные клетки по 1 в поле зрения, частично с дегрануляцией. Морфологическая картина неспецифична, но больше соответствует миопатии.
При осмотре данных, свидетельствующих о наличии аутоиммунной и эндокринологической патологии, получено не было. Для исключения миодистрофии с поздним дебютом пациент был представлен на консилиум с участием невролога, после которого был направлен на медико-генетическое консультирование.
Клинический диагноз и рекомендации. Заключение медико-генетического консультирования: наследственная мышечная дистрофия Миоши. Гиперферментемия. Гомозиготная мутация ANOS. Риск для сибсов пробанда – 25%, для потомства – незначительно превышает общепопуляционный.
Рекомендовано:
1. Ограничение физических нагрузок: исключить силовые спортивные тренировки, избегать переутомления.
2. Избегать приема лекарственных препаратов из группы статинов.
3. ЭНМГ – раз в год; наблюдение невролога.
4. Электрокардиография – раз в год.
Таким образом, полученные результаты позволили диагностировать мышечную дистрофию с поздним дебютом, при которой первично выявленная гиперферментемия являлась признаком дегенеративного повреждения скелетной мускулатуры.
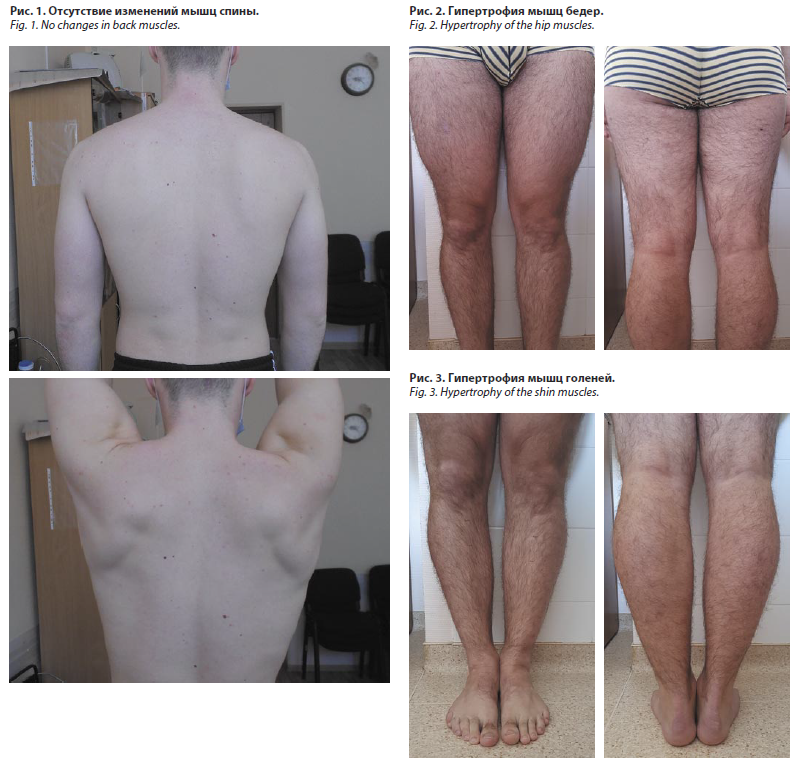
При явке в октябре 2023 г. жалоб пациент активно не предъявлял, в анализах по-прежнему сохранялись повышенные уровни трансаминаз и КФК (АЛТ – 204 Ед/л, АСТ – 235 Ед/л, КФК – 8856 Ед/л), при осмотре обращали на себя внимание компенсаторная гипертрофия мышц бедра и голени при сохраненных объеме и силе мышц спины и верхнего плечевого пояса (рис. 1–3). Диспансерная явка к неврологу была назначена на октябрь 2024 г., однако в установленный срок пациент на прием не явился.
При явке в феврале 2026 г. жалоб пациент активно не предъявляет, фитнесом не занимается. При осмотре компенсаторная гипертрофия мышц бедра и голени сохраняется на том же уровне, что и в 2023 г. В анализах сохраняются повышенные уровни трансаминаз и КФК (АЛТ – 90 Ед/л, АСТ – 86 Ед/л, КФК – 4739 Ед/л).
Обсуждение
Учитывая возраст дебюта заболевания (17 лет), отсутствие яркой клинической картины со стороны как опорно-двигательного аппарата, так и других органов и систем, а также повышение уровня КФК в 50–170 раз относительно нормальных значений, данный тип мышечной дистрофии относится к дисферлинопатиям. Поскольку дисферлинопатии могут сопровождаться повышением уровней трансаминаз внепеченочного генеза [8, 9], в ряде случаев, аналогично нашему, первоначально у таких лиц может ошибочно диагностироваться печеночная патология. На сегодняшний день описано 4 вида дисферлинопатий.
Так, для миопатии Миоши типа 1 (MMD1) характерны дебют в подростковом или раннем взрослом возрасте, ранняя и преобладающая дистальная мышечная слабость, поражающая нижние конечности, особенно икроножные и камбаловидные мышцы, поражение верхних конечностей встречается реже. Болезнь медленно прогрессирует, в начале заболевания больные не могут стоять на цыпочках и подниматься по лестнице, появляются трудности при прыжках, с годами слабость и атрофия распространяются на бедра и ягодичные мышцы, на поздних стадиях могут поражаться мышцы плеча и предплечья со снижением силы хвата, мелкие мышцы рук при этом сохранены. При дополнительном обследовании у таких лиц отмечается повышение концентрации КФК в сыворотке (в 20–150 раз выше ВГН). При биопсии атрофичных икроножных мышц обычно выявляются выраженный фиброз и замещение мышечных волокон жировой тканью [1–4].
Поясно-конечностная мышечная дистрофия рецессивного типа 2 (LGMDR2) дебютирует в подростковом возрасте или позже, прогрессирует медленно и характеризуется ранним развитием проксимальной мышечной слабости в мышцах таза. По мере прогрессирования формируется так называемая «походка дисферлина», обусловленная атрофией четырехглавой мышцы бедра, а также поражаются мышцы плечевого пояса. Отмечается массивное повышение уровня КФК в сыворотке (в 50–200 раз выше ВГН), сохраняющееся на протяжении всей болезни. С помощью компьютерной томографии (КТ) или магнитно-резонансной томографии (МРТ) можно выявить субклиническое поражение и дистальных мышц. При исследовании мышечной биопсии выявляют различные размеры мышечных волокон, увеличение соединительной ткани, жировые отложения, иногда воспаление, некроз и фиброзные изменения мышечных волокон [3, 4].
Дистальная миопатия с поражением передней большеберцовой кости (DMAT) дебютирует в подростковом возрасте, характеризуется более быстрым прогрессированием с поражением верхней части большеберцовых мышц, вызывая слабость ног и отвисание стопы, что приводит к инвалидизации в течение 10 лет от дебюта заболевания. Фиксируется умеренное повышение концентрации КФК в сыворотке крови (в 20–70 раз выше ВГН). При биопсии мышц выявляются умеренные миопатические изменения [3, 4].
Бессимптомная гиперкреатинфосфаткиназемия характеризуется выраженным повышением концентрации КФК в сыворотке крови и отсутствием явного поражения мышц. Однако данный фенотип обычно считается предсимптомным проявлением миопатии у человека, у которого в конечном итоге развиваются мышечная слабость и атрофия [3, 4].
Помимо МРТ, КТ, ЭНМГ и биопсии мышц, в диагностику дисферлинопатий входит иммуногистохимическое исследование мышечной ткани на сниженное содержание, отсутствие или аномальный дисферлин, а также генетическое тестирование на наличие мутаций гена DYSF [3, 4, 9].
Цели терапии включают сохранение подвижности и функциональной независимости пациента, лечение сопутствующих осложнений и максимальное поддержание качества жизни.
Для обеспечения оптимального лечения рекомендуется междисциплинарное лечение, включающее ортопедические пособия, физиотерапию и кинезиотерапию. В связи с тем что пациенты с мышечной дегенерацией из-за мышечной дистрофии подвержены повышенному риску повреждения мышц, вызванного физическими упражнениями, рекомендуется поддерживать адекватную гидратацию и избегать физических упражнений высокой интенсивности [3, 5–7].
В качестве экспериментальных методов лечения используются такие препараты, как эзетимиб для снижения накопления липидов в мышечной ткани; ингибиторы протеасом, потенциально снижающие провоспалительные и фибротические факторы; галектин-1 и галофугинон, потенциально стимулирующие репарацию мышечной ткани; ацетилцистеин, оказывающий антиоксидантное действие, и дилтиазем, повышающий мышечную сократимость. В качестве потенциальных методов лечения в настоящее время изучаются такие подходы, как замена и редактирование генов CRISPR/Cas9, пропуск экзонов с помощью антисмысловых олигонуклеотидов и трансплантация миобластов [3, 5–7].
Прогноз при данной патологии варьирует от длительной компенсации на уровне мышц дистальных отделов нижних конечностей до потери способности к самостоятельному передвижению. В литературе описаны случаи компенсаторных гипертрофий при миопатиях, чаще объективно выявляемых методом МРТ. В частности, для MMD1 при вовлечении в патологический процесс медиальной головки икроножной и камбаловидной мышц характерна компенсаторная гипертрофия m. sаrtоrius и m. оbturаtоrius еxtеrnus [10], что, по-видимому, и наблюдалось у нашего пациента.
Заключение
Миопатия Миоши – редкая патология с аутосомно-рецессивным типом наследования, часто проявляющаяся первоначально только повышением уровня КФК и, реже, трансаминаз, в связи с чем спектр дифференциальной диагностики представляется очень широким. Данные состояния ошибочно могут быть спутаны с множеством других патологий, начиная от сердечно-сосудистой и печеночной и заканчивая первичной патологией нервной системы.
Часто врачи первичного звена не обладают достаточными знаниями о данных заболеваниях, в связи с чем они долго остаются недиагностированными, и статистика данных случаев в Российской Федерации до сих пор неизвестна.
Конфликт интересов. Авторы заявляют об отсутствии конфликта интересов.
Conflict of interests. The authors declare no conflict of interest.
Финансирование. Исследование не имело спонсорской поддержки.
Funding. The work was performed without external funding.
Соответствие нормам этики. Участник исследования подписывал информированное добровольное согласие.
Compliance with ethical standards. The study participant signed an informed voluntary consent.
Список литературы доступен на сайте журнала https://klin-razbor.ru/
The list of references is available on the journal‘s website https://klin-razbor.ru/
Информация об авторах
Information about the authors
Полетаева Нина Борисовна – канд. мед. наук, доц. каф. поликлинической терапии ФГБОУ ВО УГМУ. E-mail: ninanova@mail.ru; ORCID: 0000-0001-7674-2893
Nina B. Poletaeva – Cand. Sci. (Med.), Assoc. Prof., Ural State Medical University.
E-mail: ninanova@mail.ru; ORCID: 0000-0001-7674-2893
Теплякова Ольга Вячеславовна – д-р мед. наук, проф. каф. поликлинической терапии ФГБОУ ВО УГМУ; врач-ревматолог, ООО «Медицинское объединение «Новая больница».
E-mail: oteplyakova69@gmail.com; ORCID: 0000-0003-2114-0419
Olga V. Teplyakova – Dr. Sci. (Med.), Prof., Ural State Medical University; rheumatologist,
New Hospital LLC. E-mail: oteplyakova69@gmail.com; ORCID: 0000-0003-2114-0419
Цаплина Полина Константиновна – ассистент каф. поликлинической терапии ФГБОУ
ВО УГМУ. E-mail: polina.tsaplina@yandex.ru; ORCID: 0009-0001-4915-4524
Polina K. Tsaplina – Assistant, Ural State Medical University. E-mail: polina.tsaplina@yandex.ru; ORCID: 0009-0001-4915-4524
Поступила в редакцию: 26.01.2026
Поступила после рецензирования: 11.02.2026
Принята к публикации: 19.02.2026
Received: 26.01.2026
Revised: 11.02.2026
Accepted: 19.02.2026
Клинический разбор в общей медицине №05 2026
Случай миопатии Миоши в клинической практике
Номера страниц в выпуске:133-137
Аннотация
Миопатия Миоши относится к группе редких миопатий – дисферлинопатий, в связи с чем долго остается недиагностированной и в настоящее время не имеет этиопатогенетического лечения. В статье приведен клинический случай миопатии Миоши у пациента 18 лет, у которого на протяжении 3 лет отмечалось повышение уровней креатинфосфокиназы и печеночных трансаминаз при отсутствии субъективных жалоб. Подробно описаны дополнительные методы исследования, использованные с целью дифференциальной диагностики, а также представлены данные медико-генетического консультирования.
Ключевые слова: миопатия Миоши, дисферлинопатия, гиперкреатинфосфатемия.
Для цитирования: Полетаева Н.Б., Теплякова О.В., Цаплина П.К. Случай миопатии Миоши в клинической практике. Клинический разбор в общей медицине. 2026; 7 (5): 133–137. DOI: 10.47407/kr2026.7.5.00854
Миопатия Миоши относится к группе редких миопатий – дисферлинопатий, в связи с чем долго остается недиагностированной и в настоящее время не имеет этиопатогенетического лечения. В статье приведен клинический случай миопатии Миоши у пациента 18 лет, у которого на протяжении 3 лет отмечалось повышение уровней креатинфосфокиназы и печеночных трансаминаз при отсутствии субъективных жалоб. Подробно описаны дополнительные методы исследования, использованные с целью дифференциальной диагностики, а также представлены данные медико-генетического консультирования.
Ключевые слова: миопатия Миоши, дисферлинопатия, гиперкреатинфосфатемия.
Для цитирования: Полетаева Н.Б., Теплякова О.В., Цаплина П.К. Случай миопатии Миоши в клинической практике. Клинический разбор в общей медицине. 2026; 7 (5): 133–137. DOI: 10.47407/kr2026.7.5.00854
A case of Miyoshi myopathy in clinical practice
Nina B. Poletaeva1, Olga V. Teplyakova1,2, Polina K. Tsaplina11 Ural State Medical University, Yekaterinburg, Russia;
2 New Hospital LLC, Yekaterinburg, Russia
polina.tsaplina@yandex.ru
Abstract
Miyoshi myopathy belongs to the group of dysferlinopathies, rare myopathies, and therefore remains undiagnosed for a long time and currently has no etiopathogenetic treatment. The article presents a clinical case of Miyoshi myopathy in an 18-year-old patient who had an increase in creatine phosphokinase and hepatic transaminases for 3 years in the absence of subjective complaints. The article describes in detail additional diagnostic tests carried out for the purpose of differential diagnosis, as well as presents the data of medical genetic counseling.
Keywords: Miyoshi myopathy, dysferlinopathy, hypercreatinephosphatemia.
For citation: Poletaeva N.B., Teplyakova O.V., Tsaplina P.K. A case of Miyoshi myopathy in clinical practice. Clinical review for general practice. 2026; 7 (5): 133–137 (In Russ.). DOI: 10.47407/kr2026.7.5.00854
Введение
Дисферлинопатии – группа редких, фенотипически разнородных мышечных заболеваний. В основе дисферлинопатий лежит дефект синтеза дисферлина – белка, участвующего в репарации мембран мышечных клеток, поэтому следствием нарушения его синтеза является постепенная дегенерация скелетной мускулатуры [1–3].
В настоящее время описано как минимум 9 фенотипов дисферлинопатий, однако наиболее изучены четыре: миопатия Миоши типа 1 (MMD1), поясно-конечностная мышечная дистрофия рецессивного типа 2 (LGMDR2), дистальная миопатия с поражением передней большеберцовой кости (DMAT), бессимптомная гиперкреатинфосфаткиназемия. Все подтипы характеризуются аутосомно-рецессивным типом наследования, повышением уровня сывороточной креатинфосфокиназы (КФК) и, в большинстве случаев, слабостью определенных групп мышц [3, 4].
В настоящее время прогноз пациентов с дисферлинопатиями не определен, этиопатогенетическое лечение не разработано [3, 5–7].
Клинический случай
Пациент Б., 18 лет, студент, в феврале 2021 г. направлен на первичную консультацию к ревматологу в связи с изменениями в анализах крови в виде повышения уровня КФК более чем в 120 раз от верхней границы нормы (ВГН), а также аланинаминотрансферазы (АЛТ) и аспартатаминотрансферазы (АСТ) при отсутствии каких-либо субъективных жалоб.
Анамнез заболевания. Впервые повышение КФК было выявлено в 2019 г. в 17-летнем возрасте при прохождении «спортивной комиссии», ранее подобных исследований не проводилось. Обращала на себя внимание стойкость лабораторных изменений, кроме того, одновременно определялось повышение уровней АЛТ и АСТ.
В связи с установленными лабораторными изменениями в 2020 г. пациент был направлен на дообследование в гастроэнтерологическое отделение детской городской больницы с диагнозом «криптогенный гепатит».
По данным представленной медицинской документации, были проведены следующие лабораторные исследования:
• общеклинические анализы крови и мочи, копрограмма – без патологии.
• биохимический анализ крови: общий белок – 89,4 г/л; альбумины – 58,6% (52,4 г/л); глобулины: альфа-1 – 3,5%, альфа-2 – 7,8%, бета – 10%, гамма – 19,9%; мочевина – 3,8 ммоль/л; общий холестерин – 3,8 ммоль/л.
• HBsAg и anti-HCV – не обнаружены.
• тромбоэластограмма – нормокоагуляция в цельной крови.
• концентрация меди в сыворотке крови – 23,6 мкмоль/л (норма 11–22 мкмоль/л).
• церулоплазмин – 269,3 мг/л (норма 90–510 мг/л).
• анализ суточной мочи на медь – 0,0547 мг/сут (норма 0,0150–0,05 мг/сут).
• тандемная масс-спектрометрия – данных, свидетельствующих о наличии наследственных аминоацидопатий, органических ацидурий и дефектов окисления жирных кислот, не получено.
• IgA и IgG к деамидированным пептидам глиадина, эндомизию и тканевой трансглутаминазе – в пределах нормы.
Также были проведены следующие инструментальные исследования:
• ультразвуковое исследование органов брюшной полости – органической патологии не выявлено.
• эзофагогастродуоденоскопия: умеренно выраженный гастрит тела и антрального отдела желудка, дуоденогастральный рефлюкс, произведена биопсия слизистой оболочки двенадцатиперстной кишки.
• гистологическое исследование биопсийного материала слизистой оболочки двенадцатиперстной кишки – структура слизистой оболочки сохранена, атрофии и метаплазии нет, щеточная кайма сохранена; интраэпителиальный лимфоцитоз и нейтрофилез. Заключение: морфологическая картина соответствует классификации Marsh1, не исключается целиакия в сочетании с активным дуоденитом.
• пункционная биопсия печени под ультразвуковым контролем: в гепатобиоптате явления гиалиново-капельной белковой дистрофии гепатоцитов, умеренный портальный склероз, очаговая лимфогистиоцитарная инфильтрация, занимающая менее ⅓ портальных трактов, мелкое скопление мононуклеаров в паренхиме дольки. Заключение: хронический гепатит с перипортальным склерозом, минимальной активности. Индекс гистологической активности – 3 балла. Гель-инфузионная сонография – 1 балл. Окраска на медь и железо – отрицательно.
Таким образом, полученные данные не объясняли повышение уровней КФК и трансаминаз. Пациент был выписан под наблюдение гастроэнтеролога с рекомендациями приема эзомепразола в течение 1 мес и урсодезоксихолиевой кислоты в течение 6 мес.
Далее в течение года пациент неоднократно выполнял исследования как на фоне спортивных тренировок, так и без них. Максимальные значения КФК на фоне тренировок достигали уровня 31 000 Ед/л (норма до 180 Ед/л), в случае месячного перерыва от активных физических упражнений уровень КФК снижался до 9000 Ед/л, но по-прежнему более чем в 50 раз превышал ВГН. Одновременно неоднократно диагностировалось повышение уровней АЛТ и АСТ – до 5–6-кратного превышения ВГН.
Анамнез жизни. Единственный ребенок в семье. Начал заниматься фитнесом с 16 лет. Употребление каких-либо лекарственных средств, биологически активных и спортивных добавок пациент категорически отрицает. Биохимические исследования крови у родителей не выявили отклонений от нормы.
Данные объективного осмотра (апрель 2022 г.). Состояние удовлетворительное, температура тела 36,6°C. Кожа чистая, отеков нет. Периферические лимфатические узлы не изменены, щитовидная железа не пальпируется. Аускультативно в легких везикулярное дыхание, хрипы не выслушиваются. Тоны сердца ясные, ритм правильный, шумы отсутствуют, частота сердечных сокращений – 51 в минуту. Артериальное давление на обеих руках 120/70 мм рт. ст. Живот при пальпации мягкий, безболезненный. Печень по краю реберной дуги, селезенка не пальпируется. Мочеиспускание без изменений, стул регулярный, оформленный.
Status localis: суставы не деформированы, без экссудации, движения в полном объеме. Сила дистальных и проксимальных мышц сохранена.
Дополнительные методы исследования. С целью исключения эндокринологической и аутоиммунной патологии, в частности полимиозита, были проведены исследования крови на тиреотропный гормон, паратгормон, кальций, калий и натрий – результаты в пределах нормальных значений. Иммуноблот атинуклеарных антител – отрицательный результат.
Были проведены следующие инструментальные исследования:
• стимуляционная электронейромиография (ЭНМГ) – проведение не нарушено.
• игольчатая ЭНМГ передней большеберцовой мышцы справа, vastus medianus четырехглавой мышцы слева, дельтовидной мышцы слева: выявлены потенциалы двигательных единиц повышенной амплитуды и уменьшенной длительности, что можно расценивать как признак первично-мышечной патологии в стадии компенсации. Минимальная спонтанная активность в виде потенциалов фибрилляции (+) выявлена в четырехглавой и дельтовидной мышцах, что может свидетельствовать о минимально выраженном процессе мышечной дегенерации.
• биопсия четырехглавой мышцы бедра: в биоптате фрагменты скелетной мышцы. Поперечная исчерченность в основном сохранена. Соединительнотканные прослойки (эндомизий и перимизий) с мелкими очагами базофилии и незначительной лимфогистиоцитарной фокальной инфильтрацией с наличием плазмоцитов. Единичные фокусы с проникновением клеток инфильтрата в мышечное волокно с его повреждением глыбчатым распадом. Сосуды в основном не изменены, но в стенках отдельных артериол – единичные лимфоциты. В нескольких срезах инфильтрация более выражена и в лимфатических сосудах выявляются клеточные лимфогистиоцитарные «пробки» с примесью плазмоцитов. В эндомизии в зоне более выраженной инфильтрации определяются и тучные клетки по 1 в поле зрения, частично с дегрануляцией. Морфологическая картина неспецифична, но больше соответствует миопатии.
При осмотре данных, свидетельствующих о наличии аутоиммунной и эндокринологической патологии, получено не было. Для исключения миодистрофии с поздним дебютом пациент был представлен на консилиум с участием невролога, после которого был направлен на медико-генетическое консультирование.
Клинический диагноз и рекомендации. Заключение медико-генетического консультирования: наследственная мышечная дистрофия Миоши. Гиперферментемия. Гомозиготная мутация ANOS. Риск для сибсов пробанда – 25%, для потомства – незначительно превышает общепопуляционный.
Рекомендовано:
1. Ограничение физических нагрузок: исключить силовые спортивные тренировки, избегать переутомления.
2. Избегать приема лекарственных препаратов из группы статинов.
3. ЭНМГ – раз в год; наблюдение невролога.
4. Электрокардиография – раз в год.
Таким образом, полученные результаты позволили диагностировать мышечную дистрофию с поздним дебютом, при которой первично выявленная гиперферментемия являлась признаком дегенеративного повреждения скелетной мускулатуры.
При явке в октябре 2023 г. жалоб пациент активно не предъявлял, в анализах по-прежнему сохранялись повышенные уровни трансаминаз и КФК (АЛТ – 204 Ед/л, АСТ – 235 Ед/л, КФК – 8856 Ед/л), при осмотре обращали на себя внимание компенсаторная гипертрофия мышц бедра и голени при сохраненных объеме и силе мышц спины и верхнего плечевого пояса (рис. 1–3). Диспансерная явка к неврологу была назначена на октябрь 2024 г., однако в установленный срок пациент на прием не явился.
При явке в феврале 2026 г. жалоб пациент активно не предъявляет, фитнесом не занимается. При осмотре компенсаторная гипертрофия мышц бедра и голени сохраняется на том же уровне, что и в 2023 г. В анализах сохраняются повышенные уровни трансаминаз и КФК (АЛТ – 90 Ед/л, АСТ – 86 Ед/л, КФК – 4739 Ед/л).
Обсуждение
Учитывая возраст дебюта заболевания (17 лет), отсутствие яркой клинической картины со стороны как опорно-двигательного аппарата, так и других органов и систем, а также повышение уровня КФК в 50–170 раз относительно нормальных значений, данный тип мышечной дистрофии относится к дисферлинопатиям. Поскольку дисферлинопатии могут сопровождаться повышением уровней трансаминаз внепеченочного генеза [8, 9], в ряде случаев, аналогично нашему, первоначально у таких лиц может ошибочно диагностироваться печеночная патология. На сегодняшний день описано 4 вида дисферлинопатий.
Так, для миопатии Миоши типа 1 (MMD1) характерны дебют в подростковом или раннем взрослом возрасте, ранняя и преобладающая дистальная мышечная слабость, поражающая нижние конечности, особенно икроножные и камбаловидные мышцы, поражение верхних конечностей встречается реже. Болезнь медленно прогрессирует, в начале заболевания больные не могут стоять на цыпочках и подниматься по лестнице, появляются трудности при прыжках, с годами слабость и атрофия распространяются на бедра и ягодичные мышцы, на поздних стадиях могут поражаться мышцы плеча и предплечья со снижением силы хвата, мелкие мышцы рук при этом сохранены. При дополнительном обследовании у таких лиц отмечается повышение концентрации КФК в сыворотке (в 20–150 раз выше ВГН). При биопсии атрофичных икроножных мышц обычно выявляются выраженный фиброз и замещение мышечных волокон жировой тканью [1–4].
Поясно-конечностная мышечная дистрофия рецессивного типа 2 (LGMDR2) дебютирует в подростковом возрасте или позже, прогрессирует медленно и характеризуется ранним развитием проксимальной мышечной слабости в мышцах таза. По мере прогрессирования формируется так называемая «походка дисферлина», обусловленная атрофией четырехглавой мышцы бедра, а также поражаются мышцы плечевого пояса. Отмечается массивное повышение уровня КФК в сыворотке (в 50–200 раз выше ВГН), сохраняющееся на протяжении всей болезни. С помощью компьютерной томографии (КТ) или магнитно-резонансной томографии (МРТ) можно выявить субклиническое поражение и дистальных мышц. При исследовании мышечной биопсии выявляют различные размеры мышечных волокон, увеличение соединительной ткани, жировые отложения, иногда воспаление, некроз и фиброзные изменения мышечных волокон [3, 4].
Дистальная миопатия с поражением передней большеберцовой кости (DMAT) дебютирует в подростковом возрасте, характеризуется более быстрым прогрессированием с поражением верхней части большеберцовых мышц, вызывая слабость ног и отвисание стопы, что приводит к инвалидизации в течение 10 лет от дебюта заболевания. Фиксируется умеренное повышение концентрации КФК в сыворотке крови (в 20–70 раз выше ВГН). При биопсии мышц выявляются умеренные миопатические изменения [3, 4].
Бессимптомная гиперкреатинфосфаткиназемия характеризуется выраженным повышением концентрации КФК в сыворотке крови и отсутствием явного поражения мышц. Однако данный фенотип обычно считается предсимптомным проявлением миопатии у человека, у которого в конечном итоге развиваются мышечная слабость и атрофия [3, 4].
Помимо МРТ, КТ, ЭНМГ и биопсии мышц, в диагностику дисферлинопатий входит иммуногистохимическое исследование мышечной ткани на сниженное содержание, отсутствие или аномальный дисферлин, а также генетическое тестирование на наличие мутаций гена DYSF [3, 4, 9].
Цели терапии включают сохранение подвижности и функциональной независимости пациента, лечение сопутствующих осложнений и максимальное поддержание качества жизни.
Для обеспечения оптимального лечения рекомендуется междисциплинарное лечение, включающее ортопедические пособия, физиотерапию и кинезиотерапию. В связи с тем что пациенты с мышечной дегенерацией из-за мышечной дистрофии подвержены повышенному риску повреждения мышц, вызванного физическими упражнениями, рекомендуется поддерживать адекватную гидратацию и избегать физических упражнений высокой интенсивности [3, 5–7].
В качестве экспериментальных методов лечения используются такие препараты, как эзетимиб для снижения накопления липидов в мышечной ткани; ингибиторы протеасом, потенциально снижающие провоспалительные и фибротические факторы; галектин-1 и галофугинон, потенциально стимулирующие репарацию мышечной ткани; ацетилцистеин, оказывающий антиоксидантное действие, и дилтиазем, повышающий мышечную сократимость. В качестве потенциальных методов лечения в настоящее время изучаются такие подходы, как замена и редактирование генов CRISPR/Cas9, пропуск экзонов с помощью антисмысловых олигонуклеотидов и трансплантация миобластов [3, 5–7].
Прогноз при данной патологии варьирует от длительной компенсации на уровне мышц дистальных отделов нижних конечностей до потери способности к самостоятельному передвижению. В литературе описаны случаи компенсаторных гипертрофий при миопатиях, чаще объективно выявляемых методом МРТ. В частности, для MMD1 при вовлечении в патологический процесс медиальной головки икроножной и камбаловидной мышц характерна компенсаторная гипертрофия m. sаrtоrius и m. оbturаtоrius еxtеrnus [10], что, по-видимому, и наблюдалось у нашего пациента.
Заключение
Миопатия Миоши – редкая патология с аутосомно-рецессивным типом наследования, часто проявляющаяся первоначально только повышением уровня КФК и, реже, трансаминаз, в связи с чем спектр дифференциальной диагностики представляется очень широким. Данные состояния ошибочно могут быть спутаны с множеством других патологий, начиная от сердечно-сосудистой и печеночной и заканчивая первичной патологией нервной системы.
Часто врачи первичного звена не обладают достаточными знаниями о данных заболеваниях, в связи с чем они долго остаются недиагностированными, и статистика данных случаев в Российской Федерации до сих пор неизвестна.
Конфликт интересов. Авторы заявляют об отсутствии конфликта интересов.
Conflict of interests. The authors declare no conflict of interest.
Финансирование. Исследование не имело спонсорской поддержки.
Funding. The work was performed without external funding.
Соответствие нормам этики. Участник исследования подписывал информированное добровольное согласие.
Compliance with ethical standards. The study participant signed an informed voluntary consent.
Список литературы доступен на сайте журнала https://klin-razbor.ru/
The list of references is available on the journal‘s website https://klin-razbor.ru/
Информация об авторах
Information about the authors
Полетаева Нина Борисовна – канд. мед. наук, доц. каф. поликлинической терапии ФГБОУ ВО УГМУ. E-mail: ninanova@mail.ru; ORCID: 0000-0001-7674-2893
Nina B. Poletaeva – Cand. Sci. (Med.), Assoc. Prof., Ural State Medical University.
E-mail: ninanova@mail.ru; ORCID: 0000-0001-7674-2893
Теплякова Ольга Вячеславовна – д-р мед. наук, проф. каф. поликлинической терапии ФГБОУ ВО УГМУ; врач-ревматолог, ООО «Медицинское объединение «Новая больница».
E-mail: oteplyakova69@gmail.com; ORCID: 0000-0003-2114-0419
Olga V. Teplyakova – Dr. Sci. (Med.), Prof., Ural State Medical University; rheumatologist,
New Hospital LLC. E-mail: oteplyakova69@gmail.com; ORCID: 0000-0003-2114-0419
Цаплина Полина Константиновна – ассистент каф. поликлинической терапии ФГБОУ
ВО УГМУ. E-mail: polina.tsaplina@yandex.ru; ORCID: 0009-0001-4915-4524
Polina K. Tsaplina – Assistant, Ural State Medical University. E-mail: polina.tsaplina@yandex.ru; ORCID: 0009-0001-4915-4524
Поступила в редакцию: 26.01.2026
Поступила после рецензирования: 11.02.2026
Принята к публикации: 19.02.2026
Received: 26.01.2026
Revised: 11.02.2026
Accepted: 19.02.2026
Список исп. литературыСкрыть список1. Moore U, Gordish H, Diaz-Manera J et al. Miyoshi myopathy and limb girdle muscular dystrophy R2 are the same disease. Neuromuscul Disord 2021 Apr;31(4):265-80. DOI: 10.1016/j.nmd.2021.01.009
2. Stolarski L, Patrzalek P, Gerber F et al. Clinical Presentation, Diagnosis, and Genetic Insights of Miyoshi Myopathy: A Case Report and Literature Review. Cureus 2024 Sep 7;16(9):e68869. DOI: 10.7759/cureus. 68869
3. Anwar S, Yokota T. The Dysferlinopathies Conundrum: Clinical Spectra, Disease Mechanism and Genetic Approaches for Treatments. Biomolecules 2024 Feb 21;14(3):256. DOI: 10.3390/biom14030256
4. Ivanova A, Smirnikhina S, Lavrov A. Dysferlinopathies: Clinical and genetic variability. Clin Genet 2022 Dec;102(6):465-73. DOI: 10.1111/cge. 14216
5. Poudel BH, Fletcher S, Wilton SD, Aung-Htut M. Limb Girdle Muscular Dystrophy Type 2B (LGMD2B): Diagnosis and Therapeutic Possibilities. Int J Mol Sci 2024 May 21;25(11):5572. DOI: 10.3390/ ijms25115572
6. Moore U, Jacobs M, Fernandez-Torron R et al. Intensive Teenage Activity Is Associated With Greater Muscle Hyperintensity on T1W Magnetic Resonance Imaging in Adults With Dysferlinopathy. Front Neurol 2020 Dec 16;11:613446. DOI: 10.3389/fneur.2020.613446
7. Bouchard C, Tremblay JP. Portrait of Dysferlinopathy: Diagnosis and Development of Therapy. J Clin Med 2023 Sep 16;12(18):6011. DOI: 10.3390/jcm12186011
8. Федотов В.П., Рыжкова О.П., Поляков А.В. Миопатия Миоши: диагностика семейного случая дисферлинопатии. Нервно-мышечные болезни. 2016;(6):82-8.
Fedotov V.P., Ryzhkova O.P., Polyakov A.V. Miyoshi myopathy: diagnosis of a familial case of dysferlinopathy. Neuromuscular Diseases. 2016;(6):82-8 (in Russian).
9. Черданцева Т.М., Баковецкая О.В., Никифоров А.А., Некрасова М.С. Морфологические и лабораторно-генетические исследования мышечных дистрофий. Наука молодых (Eruditio Juvenium). 2021;(9):481-91. DOI: 10.23888/HMJ202193481-491
Cherdantseva T.M., Bakovetskaya O.V., Nikiforov A.A., Nekrasovа M.S. Morphological and laboratory genetic studies of muscular dystrophies. Science of the young (Eruditio Juvenium). 2021;9(3):481-91. DOI: 10.23888/HMJ202193481-491 (in Russian).
10. Царгуш В.А., Бардаков С.Н., Багненко С.С. и др. Магнитно-резонансный паттерн изменений мышц тазового пояса и нижних конечностей у пациентов с дисферлинопатиями. Лучевая диагностика и лучевая терапия. 2020;(1):93-105. DOI: 10.22328/2079-5343-2020-11-1-93-105
Tsargush V.A., Bardakov S.N., Bagnenko S.S. et al. MRI pattern changes in pelvic muscle and lower limb in patients with dysferlinopathy. Diagnostic radiology and radiotherapy. 2020;11(1):93-105. DOI: 10.22328/2079-5343-2020-11-1-93-105 (in Russian).
1 мая 2026
Количество просмотров: 43